Preprocessing¶
10X Genomics Data Files¶
Example Using a 10X Genomics Dataset¶
Output from the 10X Genomics Cell Ranger pipeline includes multiple files. The two necessary files for importing the results into tcrdist2 are:
- The Filtered contig annotations (CSV)
- The Clonotype consensus annotations (CSV)
The required fields for these file are shown at the bottom of this page. below. All data downloaded from 10X Genomics Example Datasets .
Tip
We use the following files in the coding example:
- vdj_v1_hs_pbmc_t_filtered_contig_annotations.csv
- vdj_v1_hs_pbmc_t_consensus_annotations.csv
Pre-Processing the 10X Data Files¶
Some pre-processing of the data is required to create a tcrdist2
compatible clone file from the 10X Cell Ranger files. This is done using
tcrdist.preprocess_10X.get_10X_clones().
import pandas as pd
import numpy as np
import multiprocessing
import tcrdist as td
from tcrdist import preprocess_10X
from tcrdist.repertoire import TCRrep
from tcrdist.vis_tools import bostock_cat_colors, cluster_viz
# filtered_contig_annotations_csvfile
fca = 'vdj_v1_hs_pbmc_t_filtered_contig_annotations.csv'
ca = 'vdj_v1_hs_pbmc_t_consensus_annotations.csv'
clones_df = preprocess_10X.get_10X_clones(organism = 'human',
filtered_contig_annotations_csvfile = fca,
consensus_annotations_csvfile = ca)
clones_df.head()
Tip
Multiple productive alpha or beta chains associate with a single 10X unique cell identifier are possible. tcrdist2 picks the productive variant of the chain associated with the highest number of unique molecular identifies.
Initialize TCRrep to Calculate tcrdistances¶
tr = TCRrep(cell_df = clones_df, organism = "human", chains = ["alpha","beta"])
tr.infer_cdrs_from_v_gene(chain = 'alpha', imgt_aligned=True)
tr.infer_cdrs_from_v_gene(chain = 'beta', imgt_aligned=True)
tr.index_cols = ['clone_id', 'subject',
'v_a_gene', 'j_a_gene', 'v_b_gene', 'j_b_gene',
'cdr3_a_aa', 'cdr3_b_aa',
'cdr1_a_aa', 'cdr2_a_aa', 'pmhc_a_aa',
'cdr1_b_aa', 'cdr2_b_aa', 'pmhc_b_aa',
'cdr3_b_nucseq', 'cdr3_a_nucseq']#,
#'va_countreps', 'ja_countreps',#
#'vb_countreps', 'jb_countreps',
#'va_gene', 'vb_gene',
#'ja_gene', 'jb_gene']
tr.deduplicate()
cpus = multiprocessing.cpu_count()
tr._tcrdist_legacy_method_alpha_beta(processes = cpus)
tr.clone_df['epitope'] = "Unk" # Assign an unknown epitope to all clones
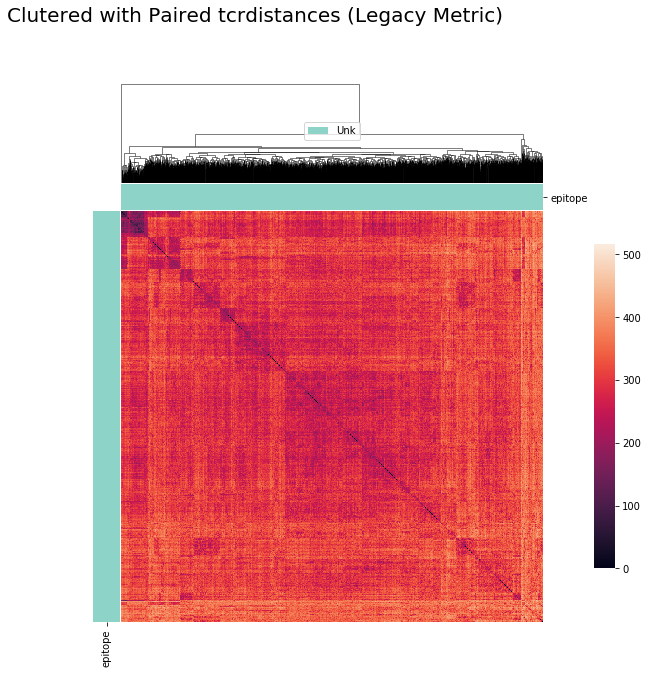
cluster_viz(pd.DataFrame(tr.paired_tcrdist),
tr.clone_df,
tr.clone_df['epitope'].unique(),
bostock_cat_colors(['set3']),
"Clutered with Paired tcrdistances (Legacy Metric)")
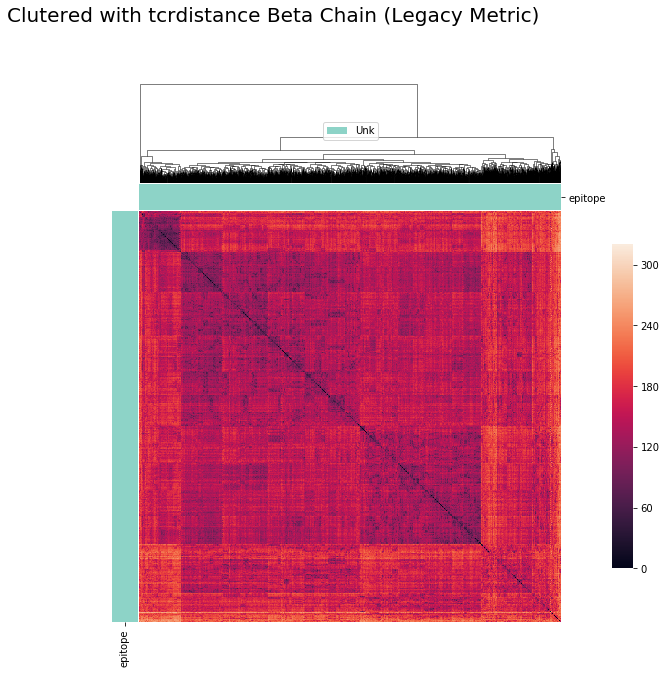
cluster_viz(pd.DataFrame(tr.dist_b.values),
tr.clone_df,
tr.clone_df['epitope'].unique(),
bostock_cat_colors(['set3']),
"Clutered with tcrdistance Beta Chain (Legacy Metric)")
Bulk Data Processed with MiXCR¶
If you have bulk sequencing data, MiXCR is an excellent option for rapidly annotating the CDRs and identifying gene V and J gene usage. Documentation for MiXCR can be found at mixcr.readthedocs.io/.
Adaptive Data File¶
Describe preprocessing of Adaptive Files
VDJDB Data Files¶
Describe Preprocessing VDJDB files
Clones File¶
Describe how to use a clones file from previous TCRdist run.
Mappers¶
The typical workflow for getting data into tcrdist2 is to load a data file with Pandas and then apply a mapper to select and rename columns to match those required by tcrdist2. see Mappers
We have begun writing wrappers for various data sources. Mappers are just python dictionaries and can be easily emulated if your data headers are unique.
import pandas as pd
from tcrdist import mappers
mapping = mappers.tcrdist_clone_df_to_tcrdist2_mapping
tcrdist1_df = mappers.generic_pandas_mapper(df = tcrdist_clone_df_PA,
mapping = mapping)
Input Files¶
10X Genomics Data Fields¶
- Required Fields in the 10X Genomics contig_annotations_csvfile
FIELD EXAMPLE VALUE barcode AAAGATGGTCTTCTCG-1 is_cell TRUE contig_id AAAGATGGTCTTCTCG-1_contig_1 high_confidence TRUE length 695 chain TRB v_gene TRBV5-1*01 d_gene TRBD2*02 j_gene TRBJ2-3*01 c_gene TRBC2*01 full_length TRUE productive TRUE cdr3 CASSPLAGYAADTQYF cdr3_nt TGCGCCAGCAGCCCCCTAGCGGGATACGCAGCAGATACGCAGTATTTT reads 9427 umis 9 raw_clonotype_id clonotype14 raw_consensus_id clonotype14_consensus_1 - Required Fields in in the 10X Genomics consensus_annotations_csvfile
FIELD EXAMPLE VALUE clonotype_id clonotype100 consensus_id clonotype100_consensus_1 length 550 chain TRB v_gene TRBV24-1*01 d_gene TRBD1*01 j_gene TRBJ2-7*01 c_gene TRBC2*01 full_length TRUE productive TRUE cdr3 CATSDPGQGGYEQYF cdr3_nt TGTGCCACCAGTGACCCCGGACAGGGAGGATACGAGCAGTACTTC reads 8957 umis 9
Adaptive Biotech Files¶
See the 3.4.13 SAMPLE EXPORT section of immunoSEQ Analyzer Manual
The standard output is expected to have column headers as shown in the table below.
- immunoSEQ
Reference Manual Definition Example1 Example2 nucleotide Full length nucleotide sequence GAGTCGCCCAGA ATCCAGCCCTCAGA aminoAcid CDR3 amino acid sequence CASSLSWYNTGELFF CASSLGQTNTEAFF count The count of sequence reads for the given nucleotide sequence 13 2 frequencyCount The percentage of a particular nucleotide sequence in the total sample Total length of the CDR3 region 0.003421953 5.26E-04 cdr3Length The highest degree of specificity obtained when identifying the V gene Name of the V family (if identified) 45 42 vMaxResolved Name of the V gene if identified, otherwise ‘unresolved’ TCRBV27-01*01 TCRBV12 vFamilyName Number of the V gene allele if identified, otherwise ‘1’ TCRBV27 TCRBV12 vGeneName Name of the potential V families for a sequence if V family identity is unresolved TCRBV27-01 vGeneAllele Name of the potential V genes for a sequence if V gene identity is unresolved 1 vFamilyTies Name of the potential V gene alleles for a sequence if V allele identity is unresolved vGeneNameTies The highest degree of specificity obtained when identifying the D gene Name of the D family (if identified) TCRBV12-03,TCRBV12-04 vGeneAlleleTies Name of the D gene if identified, otherwise ‘unresolved’ dMaxResolved Number of the D gene allele if identified, otherwise ‘1’ or ‘unresolved’ if D gene is unresolved TCRBD01-01*01 dFamilyName Name of the potential D families for a sequence if D family identity is unresolved TCRBD01 dGeneName Name of the potential D genes for a sequence if D gene identity is unresolved TCRBD01-01 dGeneAllele Name of the potential D gene alleles for a sequence if D allele identity is unresolved 1 dFamilyTies The highest degree of specificity obtained when identifying the J gene Name of the J family (if identified) TCRBD01,TCRBD02 dGeneNameTies Name of the J gene if identified, otherwise ‘unresolved’ TCRBD01-01,TCRBD02-01 dGeneAlleleTies Number of the J gene allele if identified, otherwise ‘1’ jMaxResolved Name of the potential J families for a sequence if J family identity is unresolved TCRBJ02-02*01 TCRBJ01-01*01 jFamilyName Name of the potential J genes for a sequence if J gene identity is unresolved TCRBJ02 TCRBJ01 jGeneName Name of the J gene if identified, otherwise ‘unresolved’ TCRBJ02-02 TCRBJ01-01 jGeneAllele Number of the J gene allele if identified, otherwise ‘1’ 1 1 jFamilyTies Name of the potential J families for a sequence if J family identity is unresolved jGeneNameTies Name of the potential J genes for a sequence if J gene identity is unresolved jGeneAlleleTies Name of the potential J gene alleles for a sequence if J allele identity is unresolved vDeletion The number of deletions from the 3’ end of the V segment 0 6 n1Insertion The number of insertions in the N1 region 2 4 d5Deletion The number of deletions from the 5’ end of the D segment 0 1 d3Deletion The number of deletions from the 3’ end of the D segment 10 5 n2Insertion The number of insertions in the N2 region 3 2 jDeletion The number of deletions from the 5’ end of the J segment 2 1 vIndex Distance from the start of the sequence (designated 0) to the conserved cysteine residue in the V motif 36 39 n1Index Distance from 0 to the start of the N1 53 50 dIndex Distance from 0 to the start of the D region 55 54 n2Index Distance from 0 to the start of the N2 region 57 60 jIndex Distance from 0 to the start of the J region 60 62 estimatedNumberGenomes Estimated number of T/B cell genomes present in the sample 13 2 sequenceStatus Whether the nucleotide sequence generates a functional amino acid sequence In In cloneResolved The gene segments used to construct the CDR3 (VDJ, VJ, DJ) VDJ VDJ vOrphon Not implemented at this time dOrphon Not implemented at this time jOrphon Not implemented at this time vFunction Not implemented at this time dFunction Not implemented at this time jFunction Not implemented at this time