Quick-Start Example¶
The Quick-Start Example illustrates some of the flexibility of tcrdist2.
See the Detailed Example for similar material with more complete explanations of the step-wise procedure and TCRrep class.
import pandas as pd
import numpy as np
import parasail
import matplotlib
import matplotlib.pyplot as plt
import seaborn as sns
%matplotlib inline
import tcrdist as td
from tcrdist import mappers
from tcrdist.repertoire import TCRrep
from tcrdist.vis_tools import bostock_cat_colors, cluster_viz
Preliminary Steps¶
Load, Format, and Subset the Data¶
pd_df = pd.read_csv("vdjDB_PMID28636592.tsv", sep = "\t") # 1
t_df = td.mappers.vdjdb_to_tcrdist2(pd_df = pd_df) # 2
t_df.organism.value_counts # 3
index_mus = t_df.organism == "MusMusculus" # 4
t_df_mus = t_df.loc[index_mus,:].copy() # 5
- Read the .tsv data file into a pandas DataFrame.
- Call tcrdist.mappers.vdjdb_to_tcrdist2() to select and rename the appropriate columns.
- The data has both human and mouse TCRs, which can’t be combined in a single repertoire.
- Index the sequences that come from MusMusculus (mouse).
- Create a copy of the subset DataFrame t_df, including only mouse TCRs: t_df_mus.
tr1 - Initialize the Repertoire¶
tr1 = TCRrep(cell_df = t_df_mus, organism = "mouse") # 6
tr1.infer_cdrs_from_v_gene(chain = 'alpha') # 7
tr1.infer_cdrs_from_v_gene(chain = 'beta') # 8
tr1.index_cols =['epitope', # 9
'subject',
'cdr3_a_aa',
'cdr1_a_aa',
'cdr2_a_aa',
'pmhc_a_aa',
'cdr3_b_aa',
'cdr1_b_aa',
'cdr2_b_aa',
'pmhc_b_aa']
tr1.deduplicate() # 10
tcrdist.repertoire.TCRrep for <Your TCR Repertoire Project>
with index_cols: ['epitope', 'subject', 'cdr3_a_aa', 'cdr1_a_aa', 'cdr2_a_aa', 'pmhc_a_aa', 'cdr3_b_aa', 'cdr1_b_aa', 'cdr2_b_aa', 'pmhc_b_aa']
with model organism: mouse
Detailed explanations of the steps shown above can be found in Example 1
- Create an instance of the tcrdist.repertoire.TCRrep class initialized with the t_df_mus DataFrame.
- Use tcrdist.repertoire.TCRrep.infer_cdrs_from_v_gene() to populate CDR1, CDR2 and pMHC loop fields.
- Repeat step 7, with chain set to ‘beta’.
- Specify index columns. Any sequence identical across all the index columns will be grouped at the following step. The count field keeps track of the number of identical clones (which may occur during clonal expansion)
- Call tcrdist.repertoire.TCRrep.deduplicate() to remove duplicates and create the tcrdist.repertoire.TCRrep.clone_df DataFrame.
Compute Hamming Distance Based Distances¶
tr1.compute_pairwise_all(chain = "alpha", # 11
metric = "hamming",
processes = 6,
matrix = parasail.blosum62)
tr1.compute_pairwise_all(chain = "beta", # 12
metric = "hamming",
processes = 6,
matrix = parasail.blosum62)
- with metric argument is set to either ‘hamming’, ‘nw’ or ‘custom’, tcrdist2 uses python’s multiprocessing package to parallelize pairwise distance computation.
- Repeat the previous step setting chain argument to ‘beta’.
How to Calculate a tcrdist¶
Once region based pairwise comparison have been generated with
compute_pairwise_all( ), there are two ways to compute a tcrdist
with user defined weights.
Method 1. One can simply access individual pairwise matrices after
compute_pairwise_all( ). Because the dimensions are row order in
clone_df. Individual pairewise matrices from different regions can be
combined, and weighted:
# tcrdist1 = (cdr3_alpha)
tcrdist1 = tr1.cdr3_a_aa_pw
# tcrdist2 = 3*(cdr3_alpha) + 3*(cdr4_beta)
tcrdist2 = 3 * tr1.cdr3_a_aa_pw + 3 * trd.cdr3_b_aa_pw
# tcrdist3 3*(cdr3_alpha) + 3*(cdr4_beta) + 1*(cdr1_alpha) + 1*(cdr1_beta)
tcrdist3 = 3 * tr1.cdr3_a_aa_pw + 3 * trd.cdr3_b_aa_pw + 1 * trd.cdr1_a_aa_pw + 1 * trd.cdr1_b_aa_pw
Method 2. Alternatively use the builtin function
.compute_paired_tcrdist( ) with a dictionary of weights:
# tcrdist1
weights1 =
{'cdr3_a_aa_pw': 3,'cdr3_b_aa_pw': 0,
'cdr2_a_aa_pw': 0,'cdr2_b_aa_pw': 0,
'cdr1_a_aa_pw': 0,'cdr1_b_aa_pw': 0,
'pmhc_a_aa_pw': 0,'pmhc_b_aa_pw': 0}
# or
# tcrdist2
weights2 =
{'cdr3_a_aa_pw': 3,'cdr3_b_aa_pw': 3,
'cdr2_a_aa_pw': 0,'cdr2_b_aa_pw': 0,
'cdr1_a_aa_pw': 0,'cdr1_b_aa_pw': 0,
'pmhc_a_aa_pw': 0,'pmhc_b_aa_pw': 0}
# or
# tcrdist3
weights3 =
{'cdr3_a_aa_pw': 3,'cdr3_b_aa_pw': 0,
'cdr2_a_aa_pw': 0,'cdr2_b_aa_pw': 0,
'cdr1_a_aa_pw': 1,'cdr1_b_aa_pw': 1,
'pmhc_a_aa_pw': 0,'pmhc_b_aa_pw': 0}
tcrdist1 = tr.compute_paired_tcrdist(replacement_weights = weights1, store_result = True)
tcrdist2 = tr.compute_paired_tcrdist(replacement_weights = weights2, store_result = True)
tcrdist3 = tr.compute_paired_tcrdist(replacement_weights = weights3, store_result = True)
When using the builtin method (store_result = True) the tcrdist matrices and weights can be accessed later:
tr.stored_tcrdist[-3]
tr.stored_tcrdist[-2]
tr.stored_tcrdist[-1]
Hamming Distance Based tcrdists¶
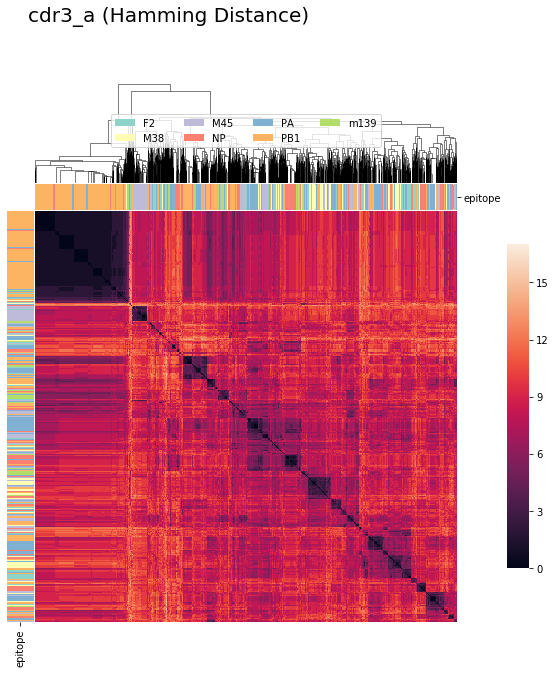
tcrdist : CDR3_alpha (Hamming Distance)¶
tcrdist1a = pd.DataFrame(tr1.cdr3_a_aa_pw)
cluster_viz(tcrdist1a,
tr1.clone_df,
tr1.clone_df.epitope.unique(),
bostock_cat_colors(['set3']),
"cdr3_a (Hamming Distance)")
/Users/kmayerbl/anaconda3/envs/py36/lib/python3.6/site-packages/seaborn/matrix.py:603: ClusterWarning: scipy.cluster: The symmetric non-negative hollow observation matrix looks suspiciously like an uncondensed distance matrix
metric=self.metric)
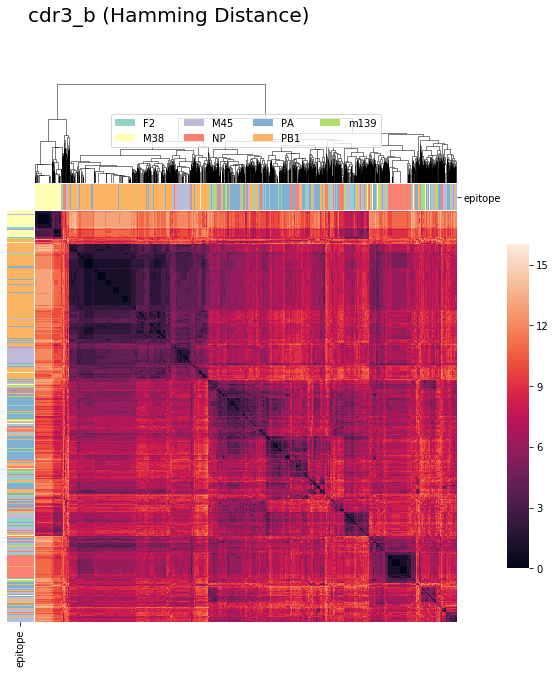
tcrdist : CDR3_beta (Hamming Distance)¶
tcrdist1b = pd.DataFrame(tr1.cdr3_b_aa_pw )
cluster_viz(tcrdist1b,
tr1.clone_df,
tr1.clone_df.epitope.unique(),
bostock_cat_colors(['set3']),
"cdr3_b (Hamming Distance)")
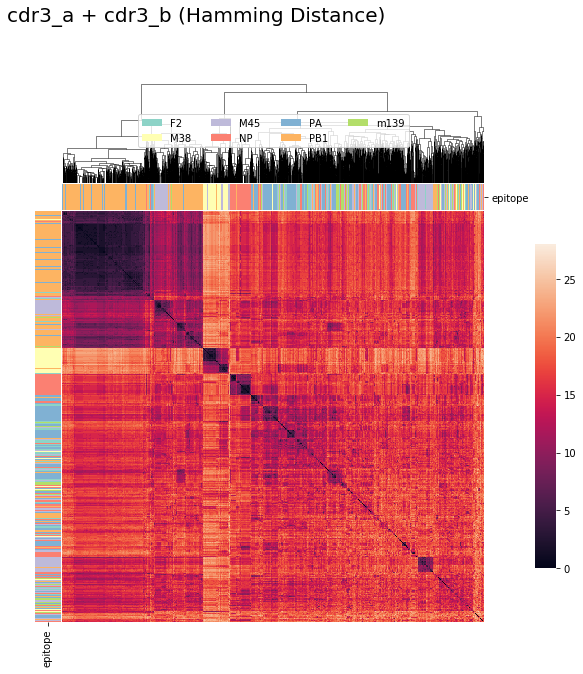
tcrdist : CDR3_alpha + CDR3_beta (Hamming Distance)¶
tcrdist2 = pd.DataFrame(tr1.cdr3_a_aa_pw + tr1.cdr3_b_aa_pw )
cluster_viz(tcrdist2,
tr1.clone_df,
tr1.clone_df.epitope.unique(),
bostock_cat_colors(['set3']),
"cdr3_a + cdr3_b (Hamming Distance)")
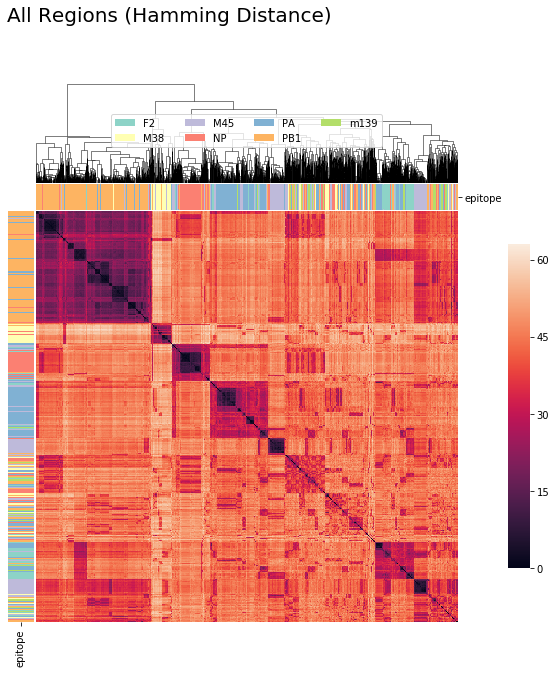
tcrdist : CDR3_alpha + CDR3_beta + Other CDR Regions (Hamming Distance)¶
tcrdist3 = pd.DataFrame(tr1.compute_paired_tcrdist(store_result= False)['paired_tcrdist'])
cluster_viz(tcrdist3,
tr1.clone_df,
tr1.clone_df.epitope.unique(),
bostock_cat_colors(['set3']),
"All Regions (Hamming Distance)")
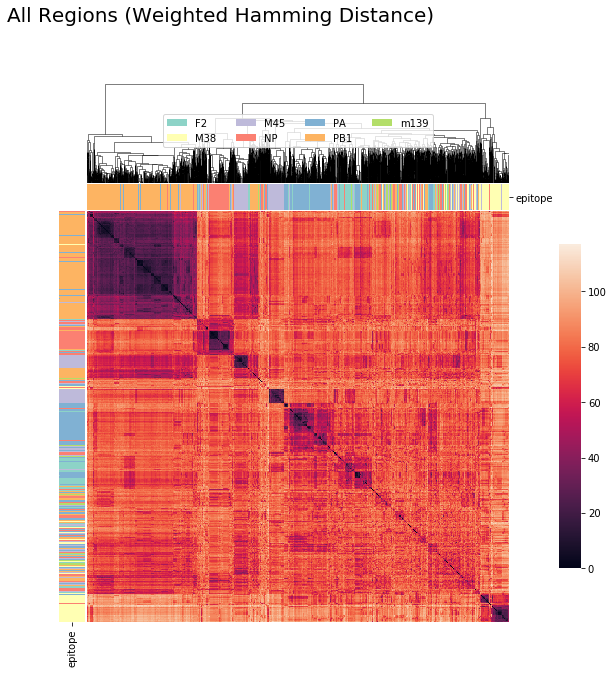
CDR3_alpha + CDR3_beta + Other CDR Regions (Weighted Hamming Distance)¶
tcrdist3w = tr1.compute_paired_tcrdist(store_result= False,
replacement_weights = {'cdr3_a_aa_pw': 3,
'cdr3_b_aa_pw': 3})
tcrdist = pd.DataFrame(tcrdist3w['paired_tcrdist'])
cluster_viz(tcrdist,
tr1.clone_df,
tr1.clone_df.epitope.unique(),
bostock_cat_colors(['set3']),
"All Regions (Weighted Hamming Distance)")
Substitution Matrix Based Distance Scores¶
It is at the .compute_pairwise_all( ) step that the choice of
distance metric is specified. When the method is specified as ‘nw’ a
reciprocal alignment score is calculated which is function of the
subsitution matrix used to score the optimal alignment (see more
explanation in example 1).
tr1.compute_pairwise_all(chain = "alpha", # 11
metric = "nw",
processes = 6,
matrix = parasail.blosum62)
tr1.compute_pairwise_all(chain = "beta", # 12
metric = "nw",
processes = 6,
matrix = parasail.blosum62)
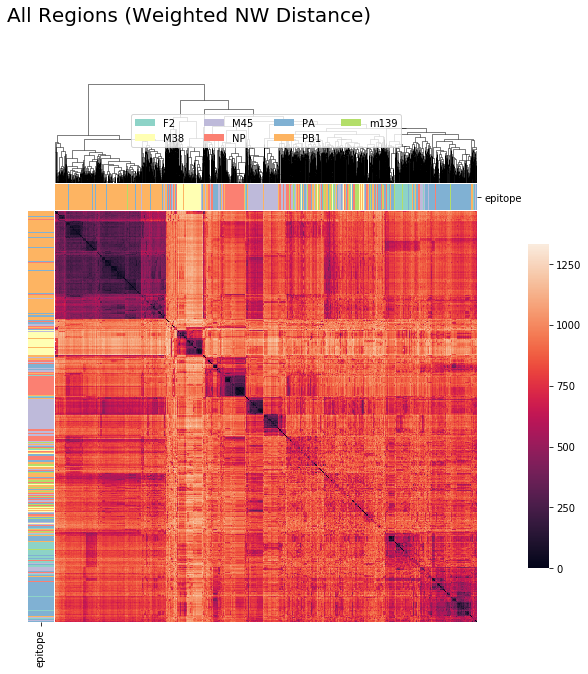
CDR3_alpha + CDR3_beta + Other CDR Regions (Weighted NW Sub Matrix Based Distance)¶
tcrdist = tr1.compute_paired_tcrdist(store_result= False,
replacement_weights = {'cdr3_a_aa_pw': 3,
'cdr3_b_aa_pw': 3})
tcrdist = pd.DataFrame(tcrdist['paired_tcrdist'])
cluster_viz(tcrdist,
tr1.clone_df,
tr1.clone_df.epitope.unique(),
bostock_cat_colors(['set3']),
"All Regions (Weighted NW Distance)")
tcrdist2 Can Parallelize Custom Metrics¶
Suppose you Imagine Some Metric¶
def hydrophobic_custom_metric(s1, s2):
s1 = s1.upper()
s2 = s2.upper()
# Types of Amino Acids
# positive_charged = ["R", "H", "K"]
# negative_charged = ["D","E"]
# polar_side_chain = ["S", "T", "N", "Q"]
# special_cases = ["C", "U", "G", "P"]
hydrophobes = ["A", "I", "L", "M", "W", "Y", "V"]
# count the number of hydrophobic amino acids in s1
h1 = np.sum([x in hydrophobes for x in list(s1)])
# count the number of hydrophobic amino acids in s2
h2 = np.sum([x in hydrophobes for x in list(s2)])
# calculate the absolute difference in hydrophobic amino acids
hydrophobic_absolute_dif = abs(h1-h2)
return int(hydrophobic_absolute_dif)
It can be passed to compute_pairwise_all( )¶
tr1.compute_pairwise_all(chain = "alpha", # 12
metric = "custom", # <----------- set metric to custom
processes = 6,
user_function = hydrophobic_custom_metric) # <----------- supply your custom function
tr1.compute_pairwise_all(chain = "beta", # 12
metric = "custom", # <----------- set metric to custom
processes = 6,
user_function = hydrophobic_custom_metric) # <----------- supply your custom function
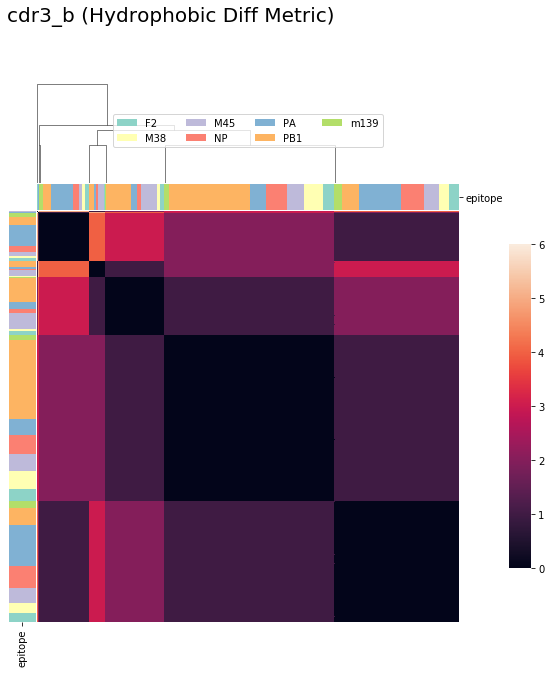
tcrdist = pd.DataFrame(tr1.cdr3_b_aa_pw)
cluster_viz(tcrdist,
tr1.clone_df,
tr1.clone_df.epitope.unique(),
bostock_cat_colors(['set3']),
"cdr3_b (Hydrophobic Diff Metric)")
tcrdist = pd.DataFrame(tr1.cdr3_a_aa_pw)
cluster_viz(tcrdist,
tr1.clone_df,
tr1.clone_df.epitope.unique(),
bostock_cat_colors(['set3']),
"cdr3_a (Hydrophobic Diff Metric)")